


SequencherDNA序列分析软件
Sequencher是一款DNA序列拼接和分析软件,它可以和自动序列分析仪一同工作,并且因为它的Conting组装、学习曲线、用户友好的编辑工具,以及技术而被周知。
Sequencher内含有多种拼接算法,还有功能的序列编辑工具,具有ORF分析、酶切位点作图、杂合子识别等分析功能。可以应用于杂合子和SNP的检测和分析,cDNA到染色体DNA的大型间隙对准,比较排序,对置信度的、ORF转换、GeneBank化导入,以及限制酶映射等等,序列比对可直接用MEGA操作,编辑更方便,比对时间稍短。
Sequencher功能
Sanger测序
Sequencher使传统的序列组装变得,同时又能控制。
修剪序列以获得质量较高的数据。您甚至可以维护修剪标准库,以使生活。
通过直观的控件,您可以为数据选择较好的算法,“汇编为参考”。而且,如果您要处理来自不同来源的多个样本,请使用“按名称组装”自动进行组装。
借助可帮助您查找和处理歧义,检查杂合子以及在数据中移动的工具,编辑数据很。
使用置信度值可帮助整理数据,质量检查和SNP检测,以提高结果的质量。
序列编辑
Sequencher为您提供DNA序列编辑工具,您需要知道这些序列正确。您可以一次查看序列的色谱图数据,也可以正向和反向查看多个对齐的色谱图。

可以的查看已对齐的数据,也可以使用Sequencher的选择工具突出显示差异较大或质量较低的区域。
序列修整
自动化的DNA测序偶尔会产生质量较差的读数,尤其是测序引物位点附近以及较长序列运行结束时。DNA文库中的克隆序列通常载体序列,polyA或无关序列。内含子和引物序列经常位于扩增的外显子序列的侧翼。除非通过修剪将其删除,否则这些伪影都会扭曲您的序列装配和下游序列分析。
Sequencher通过了易于使用但功能的工具,可帮助您修剪质量差或模棱两可的数据:
修剪末端可从测序片段末端误导性数据。
修剪向量可删除污染序列末端的于序列的数据。
修剪参考消去了延伸超出组合参考序列的序列的末端。

在执行修剪之前,Sequencher将显示建议修剪的图形表示,使您可以进一步完善条件。

如果由于修剪太严格或想要提高覆盖率而想要再修剪序列的任一端或两端恢复一定数量的碱基,则使用“批量还原修剪末端”可以做到这一点。只单击几下,您就可以将碱基还原为几千或几千个序列,并获得对序列修剪的更多控制。
序列组装
Sequencher的直观控件使您可以设置序列装配参数并在几秒钟内对其进行调整,从而可以的装配DNA片段。Sequencher会自动比较正向和反向互补序列,以组装可能的较好重叠群,因此方向如何,您都可以组装DNA序列。

将Sequencher的多功能组装工具应用于
-
比较基因变异与参考序列
-
确认载体构建
-
组装病毒和细菌基因组
-
从cDNA文库中聚簇成成千上万个序列
-
将cDNA组装成基因组序列
-
创建底图

组装参考
参考序列是的功能,是测序和序列分析的方面的核心。您是从事SNP狩猎,从事法医学,系统发育研究,医学遗传学还是人群研究,您都使用参考序列功能。
以GenBank格式导入序列,其表将应用于该序列,并将其标记为参考序列。组装到参考序列意味着您可以将您的读段与原型参考序列进行比较。
如果您要处理来自不同来源的多个样本,则甚至可以使用“按名称组装”来自动执行工作。

您甚至可以使用“参考序列”来指导去除感兴趣之外的序列或填补序列覆盖范围内的空白。
使用方差及其的报告可视化结果
多序列比对
簇状
自4.9版以来,Clustal以成为Sequencher插件家族的一部分。它是一种广泛使用的多序列比对程序,该程序通过确定一组序列上的成对比对,然后构建以近似性对序列进行分组的树状图,然后以树状图为指导进行比对。您可以使用Clusta直接从Sequencher项目窗口中比对序列。从一系列参数中进行选择以控制对齐过程。
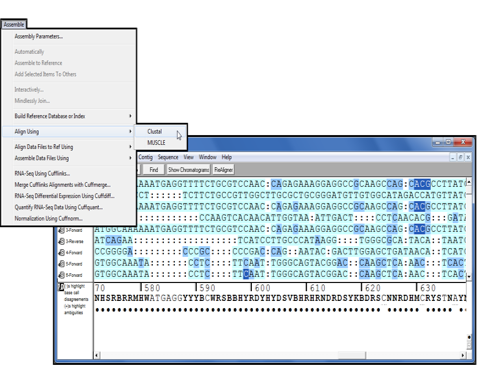
对齐完成后,您将在Sequencher中看到或多个重叠群的结果,您可以对其进行分析。
通过将Clustal于Sequencher的“按名称组装”功能相结合,可以比对来自不同来源的多个序列,从而加快Clustal的比对速度。
然后,以不同的格式(例如MSF,Phylip,NEXUS和FastA)导出结果以供程序使用,或者只是达成共识并导出。
MUSCLE
MUSCLE是多序列比对(MSA)程序,它加入了Sequencher 5.1的插件。它加入了Clustal,成为Sequencher的DNA-Seq工具中的个MSA程序。
MUSCLE在调整的过程中有四个主要步骤。初步,使用k-mer聚类在序列对之间构建距离矩阵,然后将其转换为树。步使用该数来指导渐进式对齐。在两个步骤中,MUSCLE算法尝试了多种不同的方法,以查看是否有可能并因此多重对齐方式。
一旦过程完成并且MUSCLE建立了比对,您将在Sequencher中将结果视为重叠群。使用Sequencher的工具注释比对或导出比对并将其放入的系统发育程序中。

MUSCLE是命令行程序,这意味着通常您将通过终端程序使用此程序。Sequencher使您可以使用MUSCLE的功能,而不会出现学习使用UNIX命令行的问题。
限制映射
Sequencher提供了非工具集,可用于生成DNA序列的线性限制酶切图。通过频率,突出端质或识别序列的长度来筛选您的酶选择。

您还可以指定的载体和多接头序列,以帮助您设置克隆策略。

置信度值
Sequencher在“项目”窗口,“序列编辑器”和“序列获取信息”窗口中显示置信度和汇总置信度信息(如果在DNA序列文件中可用),因此您可以的监视数据质量。

您甚至可以为您的置信度值指定截止范围,并通过颜色代码查看这些范围。

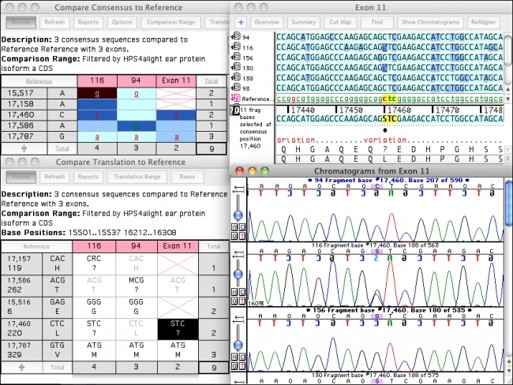
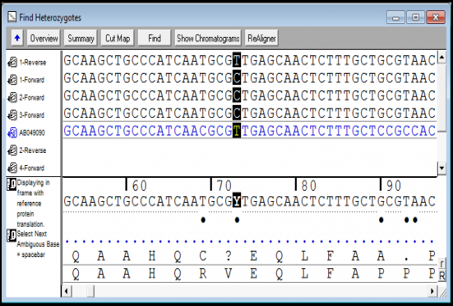
SNP检测
Sequencher有几个的工具可帮助您检测DNA序列中的突变和SNP。您可以使用Sequencher在一组序列之间进行序列比对,或将或多个序列与参考序列进行比较。Sequencher的Call Secondary Peaks...功能可分析您序列的潜在杂合子。控制定义杂合子的严格性很。

绘制DNA装配总揽中杂合子的位置图

您可以从杂合子导航到下:在“基本”视图中单击空格键即可。查看共有序列和低于共识序列的参考序列的蛋白质转变。参考序列可从DNA装配到下DNA装配,SNP的编号是的。
自动化分析
Sequencher以透明,用户可定义和可恢复的方式批量处理您的DNA序列数据,而Sequencher从来不会为了自动haul而损害科学结论。在序列编辑中,Sequencher始终为您提供选择。
Sequencher始终维护您数据的两个副本,即已编辑的数据和原始导入的数据。当您将“还原为实验数据”命令应用于项目中选定的序列或选定序列中的碱基时,可以撤销或部分编辑。
“按名称组装”工具使您可以选择片段名称的一部分,以充当共享标识符或“程序集句柄”。然后,Sequencher进行选择并自动命名重叠群。Sequencher甚至使用正则表达式匹配来设置ID!

例如,通过单击按钮,您可以将90个文件,45对正向和反向序列转换为根据您的ID命名的45个重叠群。序列装配参数的更改会重新组合片段,因此您可以根据克隆ID,日期,引物或您在序列名称中记录的来装配重叠群。

如果您进行大量测序,并且有大量样品是使用标准的测序引物完成的,则按名称组装将有用。“按名称组装”的一些应用程序可以:
-
测序MHC和线粒体基因以进行鉴定和种群研究
-
候选基因PCR产物的分析
-
跨物种对保守基因组序列并跟踪对抗病毒或疫苗的耐药性
Sequencher的常规分析
Sequencher的参考序列是功能,可控制编号,功能等。将其与方差结合使用,可以确定您是要查看已知的还是未知的SNP。
使用Clustal从Sequencher项目桌面直接比对序列。当您来自不同来源的多个样本时,甚至可以将“按名称组装”与Clustal一起使用。
根据您的工作生成报告。方差表中的PDF报告是共享信息,保存在实验室笔记本中或用于演示的好方法,
自定义工作区,控制窗口的位置,并使用标签分序列和重叠群。使用主题和突出显示来标识序列区域。在模板中维护设置,以便您和您的同事可以按照相同的标准操作程序进行工作。

自定义您的工作区
您可以定义窗口的默认位置,Sequencher会记住格式和共识选项的设置。您还可以将设置(参考序列)保存为项目模板,可以重复使用以节省分析设置时间。

-
使用主题突出显示您定义的子序列
-
记住会话之间的标尺设置
-
为您的工作风格选择较好的窗口布局并保存
-
自定义项目窗口的显示
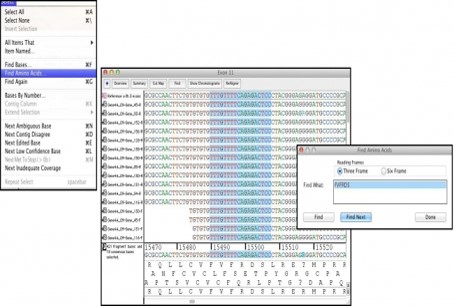
查找氨基酸
Sequencher中的“查找氨基酸”命令可以识别数据中的单个氨基酸序列。使用单字母代码,插入您要查找的氨基酸,Sequencher将在编码这些氨基酸的序列中发现并突出显示碱基。通过在“查找氨基酸”窗口中选择“查找下”按钮,Sequencher可以的找到符合搜索条件的下一组碱基。
方差表
的SNP分析工具
现在,有一种的方法可以比较DNA序列或重叠群共有序列,几次单击键就可发现这些序列之间的差异。方差表使您可以筛选大量的序列数据,从而可以可视化您感兴趣的碱基。方差表中的单元格都简介到其原始数据,因此您可以的直接在表中验证或编辑假定的差异,并自动更新序列和色谱图数据。找到差异后,Sequencher为您提供导出和报告选项。这些在“报告”页面中有更详细的描述。
多重重叠群差异表

单一重叠群差异表

翻译方差表
翻译后的差异表汇总了一组选定样品中的氨基酸差异。因为该表链接到基础序列数据,它是验证氨基酸翻译发生变化的突变以及检查表达载体的选择。

将与参考的匹配项显示为空白单元格不同,翻译后的方差表以浅灰色显示匹配项。这使您可以看到仍然匹配氨基酸的密码子差异。您甚至可以在“查看”模式下看到“变异表”和“翻译变异表”,因此您可以确定哪些DNA序列变化蛋白质序列变化。

报告书
Sequencher的输出报告可创建数据的可打印版本。报告提供了分析工具,例如人口报告中样本的距离。
人口报告是方差表中数据的摘要。人口报告均两种类型的表格;人口表和几个单独的详细表,它们描述了人口表中的组。

差异详细报告提供了Sequencher中可用的所以信息,以“差异表”。像在个别差异报告中一样,摘要表列出了样本或列的变量。此外,在差异明细报告中,描述的变式的明细表都在变式表之后。在表中的详细信息有助于调用Variant的数据的序列名称,方向和碱基调用。如果有置信度或色谱数据,则表中序列也这些数据。色谱图数据次级峰贡献的详细信息以及变体较多可六个示踪图的色谱图,尽管变体的示踪图的数量是可配置的。


-
2026-04-13
GMS 10.9 中文版正式发布 — 新增 PFAS 运移模拟与地下水能量(GWE)模块
GMS 10.9 中文版现已发布。本次更新新增 MODFLOW-USG Transport 对 PFAS 运移模拟的支持、MODFLOW 6 地下水能量(GWE)模型、UGrid 多项改进以及 MODFLOW 6 界面优化等功能,为地下水数值模拟与地热储能分析提供更多工具支持。
查看详情 >
-
2026-03-26
Origin 2026 SR1 服务更新包发布
Origin 2026 服务更新包1现已发布,适用于更新现有Origin或OriginPro 2026 SR0安装或全新安装。本次更新修正了智能填充、Excel公式、分组绘图批量操作及合并图形兼容性等多处问题,并解决了部分崩溃错误。安装后版本号将升级到10.3.0.197,用户可通过“帮助:关于Origin”确认更新完成。
查看详情 >
-
2026-03-10
GTAP数据库 V12已正式发布 - 附视频介绍
GTAP(Global Trade Analysis Project)是一个设立在美国普渡大学农业经济系的经济研究组织。该项目成立于1992年,旨在为贸易政策分析和可计算一般均衡(CGE)建模提供数据支持。全新版GTAP V12已于2026年2月正式发布,欢迎联系北京睿驰科技订购正版GTAP数据库。
查看详情 >

